E' un "fuoco amico" nel cervello la causa dell'Alzheimer? / Is the cause of Alzheimer's "friendly fire" in the brain?
E' un "fuoco amico" nel cervello la causa dell'Alzheimer? / Is the cause of Alzheimer's "friendly fire" in the brain?
Segnalato dal Dott. Giuseppe Cotellessa / Reported by Dr. Giuseppe Cotellessa

Illustrazione della proteina beta amiloide (Science Photo Library RF / AGF) / Illustration of the amyloid beta protein
Un numero crescente di studi sta evidenziando il ruolo cruciale del sistema immunitario nella demenza e nella neurodegenerazione, stimolando progetti terapeutici per ridurre l'infiammazione che, insieme agli accumuli di beta amiloide e proteine tau, rappresenta una firma caratteristica della malattia.
Il neuroscienziato Michael Heneka sa che le idee radicali richiedono dati convincenti. Nel 2010, pochissimi colleghi condividevano la sua convinzione che il sistema immunitario del cervello avesse un ruolo cruciale nella demenza. Così nel maggio di quell'anno, quando una serie di nuovi risultati fornì le prove più stringenti che avesse mai visto della sua teoria, avrebbe voluto sentirsi eccitato. E invece si sentiva nervoso.
Heneka e il suo gruppo avevano eliminato un gene chiave dell'infiammazione da un ceppo di topi che di solito sviluppa sintomi del morbo di Alzheimer. I topi modificati sembravano perfettamente sani. Superavano i test di memoria e mostravano a malapena i segni delle placche proteiche che sono un tratto distintivo della malattia. Eppure Heneka sapeva che i suoi colleghi avrebbero considerato i risultati troppo positivi per essere veri.
Persino lui era sorpreso di quanto stessero bene i topi; si sarebbe aspettato che la rimozione del gene, noto come Nlrp3, avrebbe protetto un po' il loro cervello, ma non di avvicinarsi alla prevenzione dei sintomi della demenza. "Pensavo che qualcosa fosse andato storto nella sperimentazione", dice Heneka, del Centro tedesco per le malattie neurodegenerative di Bonn. Rianalizzò i risultati più volte. Era mezzanotte passata quando alla fine concluse che potevano essere corretti.
Nei due anni successivi, ha stabilito che nulla era andato storto negli esperimenti. Insieme ai suoi colleghi, ha replicato ed elaborato i risultati. Da allora, numerosi studi hanno rafforzato l'idea di un legame tra demenza e sistema immunitario del cervello, evidenziando le cellule e i segnali coinvolti. Ma nessuno è riuscito a individuare quel legame in modo inequivocabile: il collegamento sembra sfuggente
e dinamico, in grado di cambiare con il progredire della malattia.
Heneka e il suo gruppo avevano eliminato un gene chiave dell'infiammazione da un ceppo di topi che di solito sviluppa sintomi del morbo di Alzheimer. I topi modificati sembravano perfettamente sani. Superavano i test di memoria e mostravano a malapena i segni delle placche proteiche che sono un tratto distintivo della malattia. Eppure Heneka sapeva che i suoi colleghi avrebbero considerato i risultati troppo positivi per essere veri.
Persino lui era sorpreso di quanto stessero bene i topi; si sarebbe aspettato che la rimozione del gene, noto come Nlrp3, avrebbe protetto un po' il loro cervello, ma non di avvicinarsi alla prevenzione dei sintomi della demenza. "Pensavo che qualcosa fosse andato storto nella sperimentazione", dice Heneka, del Centro tedesco per le malattie neurodegenerative di Bonn. Rianalizzò i risultati più volte. Era mezzanotte passata quando alla fine concluse che potevano essere corretti.
Nei due anni successivi, ha stabilito che nulla era andato storto negli esperimenti. Insieme ai suoi colleghi, ha replicato ed elaborato i risultati. Da allora, numerosi studi hanno rafforzato l'idea di un legame tra demenza e sistema immunitario del cervello, evidenziando le cellule e i segnali coinvolti. Ma nessuno è riuscito a individuare quel legame in modo inequivocabile: il collegamento sembra sfuggente
Tuttavia, l'idea ha suscitato l'interesse degli investitori farmaceutici, che hanno intravisto un mercato ampio e del tutto inesplorato: circa 50 milioni di persone in tutto il mondo soffrono di demenza, un numero che secondo l'Organizzazione mondiale della sanità salirà a 82 milioni entro il 2030. Quattro degli otto progetti di ricerca su farmaci sostenuti dal Dementia Consortium – un gruppo di associazioni di beneficenza e industrie farmaceutiche con sede nel Regno Unito che ha destinato 4,5 milioni di sterline (5,1 milioni di euro circa) ai progetti – sono focalizzati sull'infiammazione.
Ma ci sono degli ostacoli. Gli scienziati non concordano ancora se il sistema immunitario debba essere stimolato o inibito a seconda delle diverse fasi della malattia. E alcuni dei problemi pratici che hanno perseguitato gli studi clinici del morbo di Alzheimer – modelli imperfetti di topo e difficoltà nel reclutare i pazienti in una fase abbastanza precoce di malattia – possono affliggere anche questo nuovo approccio.
A gravare su questo settore di ricerca come nubi oscure vi è il fatto che tutti gli studi clinici sulla malattia di Alzheimer finora hanno fallito. Ma, il bioinformatico Martin Hofmann-Apitius dell'Istituto Fraunhofer per gli algoritmi e il calcolo scientifico di Sankt Augustin, specializzato nella ricerca farmaceutica, osserva che i ricercatori hanno depositato numerosi brevetti relativi agli obiettivi correlati all'infiammazione. "Presto vedremo un'ondata di studi clinici", prevede.
Intasato e gonfio
Lo psichiatra tedesco Alois Alzheimer fu il primo a descrivere i sintomi e la patologia della demenza, all'inizio del XX secolo. Guardando al microscopio il cervello di una donna del cui declino cognitivo era stato testimone, vide – e disegnò diligentemente – le placche, che ora sappiamo contenere la proteina beta amiloide, e gli ammassi fibrillari di una proteina chiamata tau che, insieme, sono la firma caratteristica della malattia.
In quelle prime rappresentazioni del tessuto cerebrale malato, Alzheimer disegnò anche la microglia, un tipo di cellula immunitaria del cervello, annidata vicino ai neuroni. "Lo stesso Alzheimer notò le cellule e le disegnò in numero abbondante associate ai neuroni", dice Heneka.
Anche se gli schizzi non stabilivano un legame più profondo tra microglia e malattia, Heneka li ricordava quando, verso la metà degli anni novanta, i legami tra l'infiammazione e l'Alzheimer cominciarono a emergere. Era stato affascinato da alcune osservazioni epidemiologiche che mostravano che le persone che assumevano alcuni farmaci antinfiammatori (per trattare l'artrite reumatoide, per esempio) sembravano a minor rischio di sviluppare il morbo di Alzheimer rispetto alla popolazione generale.
Fu incoraggiato dagli studi secondo cui la microglia si addensa attorno alle placche e alle aree di degenerazione cerebrale e le molecole infiammatorie come le citochine si concentrano nel liquido cerebrospinale dei pazienti. La maggior parte dei ricercatori ipotizzava che quelle osservazioni riflettessero una risposta passiva al danno tissutale. Ma Heneka sospettava che l'infiammazione potesse provocare attivamente la malattia.
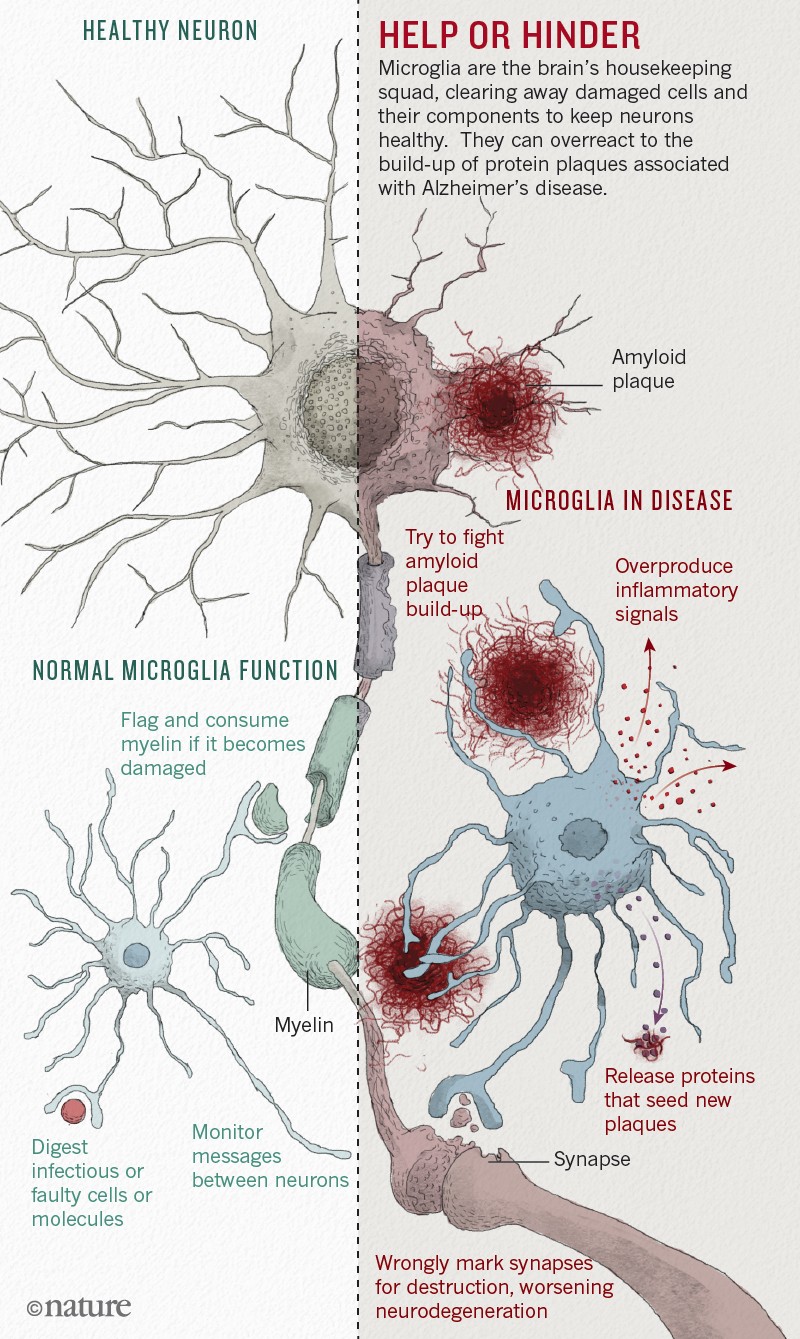
La microglia si è rivelata centrale per il legame tra infiammazione e neurodegenerazione (si veda l'infografica di "Nature").
Le sue cellule hanno due funzioni principali. Si prendono cura della salute generale dei neuroni e delle loro sinapsi, le giunzioni tra i neuroni che permettono loro di comunicare. E pattugliano il cervello, alla ricerca di minacce e problemi. Quando rilevano una molecola infettiva o aberrante come la beta amiloide – o detriti provenienti dalle cellule danneggiate – si attivano e segnalano ad altre cellule della microglia di unirsi a loro in uno sforzo di sgombero e pulizia.
Alcune proteine microgliali si riuniscono in grandi complessi chiamati inflammasomi (una componente chiave dell'infiammosoma è la proteina NLRP3 di Heneka), che producono segnali di pulizia sotto forma di molecole immunitarie attivate. Gli inflammasomi di solito diminuiscono una volta terminato il lavoro, ma nell'Alzheimer sembrano rimanere attivati, continuando a produrre molecole infiammatorie senza riuscire a completare adeguatamente la pulizia.
Ma ci sono degli ostacoli. Gli scienziati non concordano ancora se il sistema immunitario debba essere stimolato o inibito a seconda delle diverse fasi della malattia. E alcuni dei problemi pratici che hanno perseguitato gli studi clinici del morbo di Alzheimer – modelli imperfetti di topo e difficoltà nel reclutare i pazienti in una fase abbastanza precoce di malattia – possono affliggere anche questo nuovo approccio.
A gravare su questo settore di ricerca come nubi oscure vi è il fatto che tutti gli studi clinici sulla malattia di Alzheimer finora hanno fallito. Ma, il bioinformatico Martin Hofmann-Apitius dell'Istituto Fraunhofer per gli algoritmi e il calcolo scientifico di Sankt Augustin, specializzato nella ricerca farmaceutica, osserva che i ricercatori hanno depositato numerosi brevetti relativi agli obiettivi correlati all'infiammazione. "Presto vedremo un'ondata di studi clinici", prevede.
Intasato e gonfio
Lo psichiatra tedesco Alois Alzheimer fu il primo a descrivere i sintomi e la patologia della demenza, all'inizio del XX secolo. Guardando al microscopio il cervello di una donna del cui declino cognitivo era stato testimone, vide – e disegnò diligentemente – le placche, che ora sappiamo contenere la proteina beta amiloide, e gli ammassi fibrillari di una proteina chiamata tau che, insieme, sono la firma caratteristica della malattia.
In quelle prime rappresentazioni del tessuto cerebrale malato, Alzheimer disegnò anche la microglia, un tipo di cellula immunitaria del cervello, annidata vicino ai neuroni. "Lo stesso Alzheimer notò le cellule e le disegnò in numero abbondante associate ai neuroni", dice Heneka.
Anche se gli schizzi non stabilivano un legame più profondo tra microglia e malattia, Heneka li ricordava quando, verso la metà degli anni novanta, i legami tra l'infiammazione e l'Alzheimer cominciarono a emergere. Era stato affascinato da alcune osservazioni epidemiologiche che mostravano che le persone che assumevano alcuni farmaci antinfiammatori (per trattare l'artrite reumatoide, per esempio) sembravano a minor rischio di sviluppare il morbo di Alzheimer rispetto alla popolazione generale.
Fu incoraggiato dagli studi secondo cui la microglia si addensa attorno alle placche e alle aree di degenerazione cerebrale e le molecole infiammatorie come le citochine si concentrano nel liquido cerebrospinale dei pazienti. La maggior parte dei ricercatori ipotizzava che quelle osservazioni riflettessero una risposta passiva al danno tissutale. Ma Heneka sospettava che l'infiammazione potesse provocare attivamente la malattia.
La microglia si è rivelata centrale per il legame tra infiammazione e neurodegenerazione (si veda l'infografica di "Nature").
Le sue cellule hanno due funzioni principali. Si prendono cura della salute generale dei neuroni e delle loro sinapsi, le giunzioni tra i neuroni che permettono loro di comunicare. E pattugliano il cervello, alla ricerca di minacce e problemi. Quando rilevano una molecola infettiva o aberrante come la beta amiloide – o detriti provenienti dalle cellule danneggiate – si attivano e segnalano ad altre cellule della microglia di unirsi a loro in uno sforzo di sgombero e pulizia.
Alcune proteine microgliali si riuniscono in grandi complessi chiamati inflammasomi (una componente chiave dell'infiammosoma è la proteina NLRP3 di Heneka), che producono segnali di pulizia sotto forma di molecole immunitarie attivate. Gli inflammasomi di solito diminuiscono una volta terminato il lavoro, ma nell'Alzheimer sembrano rimanere attivati, continuando a produrre molecole infiammatorie senza riuscire a completare adeguatamente la pulizia.
Nel 2013, la microglia ha iniziato a comparire insistentemente nella ricerca sulla malattia di Alzheimer. Più o meno nello stesso periodo in cui il documento di Heneka mostrava che prevenire l'infiammazione allontanava la patologia di Alzheimer nei topi, il "New England Journal of Medicine" ha pubblicato due ampi studi sulle varianti geniche associate alla malattia. Entrambi gli studi collegavano il rischio di sviluppare l'Alzheimer a esordio tardivo a un gene chiamato TREM2, che produce una proteina che si trova nella membrana delle cellule microgliali.
I neuroscienziati hanno iniziato a farci caso. E lo stesso hanno fatto gli immunologi. Così è nata una comunità interdisciplinare di neuroimmunologi. "All'improvviso si sono aperte enormi opportunità", afferma la neuroscienziata Michela Matteoli dell'Università di Milano, che ora gestisce un programma di neuroscienze nel dipartimento di immunologia del vicino Istituto Humanitas, dove ha trovato un tesoro di modelli murini privi di elementi specifici del sistema immunitario, che gli immunologi non avevano mai avuto motivo di usare per studiare la funzione cerebrale. "Molti degli strumenti di cui abbiamo bisogno sono disponibili", afferma.
Buoni e cattivi
Come fa la microglia, che si è evoluta per mantenere il cervello in buono stato, a diventare una forza del male nell'Alzheimer?
L'anno scorso, Heneka e colleghi hanno pubblicato prove che suggeriscono un meccanismo plausibile per quella trasformazione, almeno nei topi. Hanno scoperto che le cellule della microglia attivate scartano i resti degli inflammasomi in piccoli gruppi chiamati granuli, e che questi granuli continuano a seminare nuovi ammassi di beta amiloide, diffondendo la malattia in tutto il cervello. "Una tempesta perfetta", dice Heneka. "La beta amiloide tossica promuove l'infiammazione, che a sua volta favorisce una maggiore tossicità della beta amiloide."
Heneka sta collaborando con l'immunologo Eicke Latz, dell'Università di Bonn, per sviluppare un farmaco in grado d'inibire la formazione dell'inflammasoma. Questo consentirebbe alla microglia di continuare gli altri ruoli importanti nella pulizia del cervello senza dover ricorrere ad altre cellule di microglia. La tempesta sarebbe tenuta a bada.
Nel 2016, Latz ha co-fondato la start-up IFM Therapeutics a Boston. La società, acquisita dalla casa farmaceutica Bristol Myers Squibb lo scorso anno, ha già alcuni farmaci candidati che impediscono la formazione di inflammasomi e la speranza di Latz e Heneka è d'iniziare le sperimentazioni cliniche nei prossimi due anni.
Nel frattempo, i neuroimmunologi di tutto il mondo stanno cercando di acquisire una comprensione più profonda della biologia della microglia, per capire se ci possano essere altri modi per progettare immunoterapie per l'Alzheimer e altre malattie neurodegenerative. Alcuni scienziati pensano che le attività salutari della microglia potrebbero essere rafforzate per eliminare la beta amiloide tossica in modo più efficiente ed evitare del tutto la tempesta.
I neuroscienziati hanno iniziato a farci caso. E lo stesso hanno fatto gli immunologi. Così è nata una comunità interdisciplinare di neuroimmunologi. "All'improvviso si sono aperte enormi opportunità", afferma la neuroscienziata Michela Matteoli dell'Università di Milano, che ora gestisce un programma di neuroscienze nel dipartimento di immunologia del vicino Istituto Humanitas, dove ha trovato un tesoro di modelli murini privi di elementi specifici del sistema immunitario, che gli immunologi non avevano mai avuto motivo di usare per studiare la funzione cerebrale. "Molti degli strumenti di cui abbiamo bisogno sono disponibili", afferma.
Buoni e cattivi
Come fa la microglia, che si è evoluta per mantenere il cervello in buono stato, a diventare una forza del male nell'Alzheimer?
L'anno scorso, Heneka e colleghi hanno pubblicato prove che suggeriscono un meccanismo plausibile per quella trasformazione, almeno nei topi. Hanno scoperto che le cellule della microglia attivate scartano i resti degli inflammasomi in piccoli gruppi chiamati granuli, e che questi granuli continuano a seminare nuovi ammassi di beta amiloide, diffondendo la malattia in tutto il cervello. "Una tempesta perfetta", dice Heneka. "La beta amiloide tossica promuove l'infiammazione, che a sua volta favorisce una maggiore tossicità della beta amiloide."
Heneka sta collaborando con l'immunologo Eicke Latz, dell'Università di Bonn, per sviluppare un farmaco in grado d'inibire la formazione dell'inflammasoma. Questo consentirebbe alla microglia di continuare gli altri ruoli importanti nella pulizia del cervello senza dover ricorrere ad altre cellule di microglia. La tempesta sarebbe tenuta a bada.
Nel 2016, Latz ha co-fondato la start-up IFM Therapeutics a Boston. La società, acquisita dalla casa farmaceutica Bristol Myers Squibb lo scorso anno, ha già alcuni farmaci candidati che impediscono la formazione di inflammasomi e la speranza di Latz e Heneka è d'iniziare le sperimentazioni cliniche nei prossimi due anni.
Nel frattempo, i neuroimmunologi di tutto il mondo stanno cercando di acquisire una comprensione più profonda della biologia della microglia, per capire se ci possano essere altri modi per progettare immunoterapie per l'Alzheimer e altre malattie neurodegenerative. Alcuni scienziati pensano che le attività salutari della microglia potrebbero essere rafforzate per eliminare la beta amiloide tossica in modo più efficiente ed evitare del tutto la tempesta.
Due studi su topi e cervelli umani post-mortem hanno dimostrato che la microglia che si addensa intorno alle placche nel cervello è un sottogruppo molto specifico. Queste cellule esprimono alcuni geni a livelli più alti o più bassi rispetto alla microglia normale, e questi schemi raccontano una storia interessante: le cellule sembrano cercare di calibrare i loro normali compiti di pulizia al fine di combattere le placche.
Alcuni di questi geni rimuovono le protezioni, o checkpoint, dalle vie che portano all'attivazione delle cellule. Altri si trovano in vie che percepiscono un danno o incoraggiano la microglia a fagocitare le molecole difettose. In ogni caso, gli schemi di espressione genica indicano che le cellule della microglia stanno incrementando le loro “pulizie di casa” per cercare di proteggere il cervello.
Le mutazioni di una dozzina circa di questi geni erano già state identificate come fattori di rischio per l'Alzheimer negli esseri umani, dice Ido Amit, immunogeneticista del Weizmann Institute of Science di Rehovot, in Israele, che ha condotto uno degli studi sugli schemi di espressione genica.
Amit dice che le cellule sono lì per una ragione e potrebbero quindi essere sfruttate. "I risultati sembravano mandarci un messaggio forte sulla biologia del sistema", afferma. Se la microglia potesse essere indotta a svolgere le sue funzioni regolari in modo più efficiente, impedendole eventuali sforzi di pulizia eccessivi, ciò potrebbe contribuire a prevenire i sintomi della malattia piuttosto che peggiorarne il decorso.
Se c'erano ancora dubbi sull'importanza della microglia nei meccanismi della demenza – se cioè svolgesse il ruolo di buono o di cattivo – questi articoli li hanno fugati. Inoltre, la microglia potrebbe anche essere indotta ad attivarsi dall'infiammazione in altre parti del corpo.
Studi epidemiologici hanno dimostrato che l'accumulo di infezioni durante la vita aumenta il rischio di deterioramento cognitivo o di demenza in età avanzata. E all'inizio di questo mese, Jonas Neher del Centro tedesco per le malattie neurodegenerative di Tubinga, e colleghi, hanno dimostrato che provocare infiammazioni nei topi iniettando molecole chiamate lipopolisaccaridi (LPS) nel loro ventre portava a cambiamenti persistenti nell'espressione genica nella microglia del cervello, anche se le molecole stesse ne rimanevano fuori. Basse dosi di LPS hanno portato a un aumento dei livelli di beta amiloide e di placche; alte dosi hanno ridotto l’accumulo.
La microglia potrebbe essere coinvolta anche in altre malattie neurodegenerative, poiché risultati simili sono stati ottenuti in modelli di sclerosi laterale amiotrofica (SLA) e morbo di Parkinson. E le ricerche di Matteoli e altri suggeriscono che potrebbero essere implicati ancora più ampiamente nei disturbi cerebrali, come il raro disturbo dello sviluppo neurologico noto come sindrome di Rett.
Al riparo dalla tempesta
Amit sta ora discutendo con i partner industriali su come potenziare le attività di pulizia della microglia. "Questo ci permetterebbe di riattivare le nostre difese naturali quando il danno è fuori controllo", dice.
Altri temono che una maggiore attivazione di microglia nelle ultime fasi della malattia possa peggiorare le cose. "Non sappiamo ancora abbastanza della biologia", dice Oleg Butovsky, neuroimmunologo della Harvard Medical School di Boston, che ha condotto l'altro studio sull'espressione genica nella microglia e sta sviluppando biomarcatori che possano essere identificati nel cervello in diverse fasi del disturbo. Egli sostiene che non è chiaro se la microglia debba essere potenziata o soppressa, o anche se tattiche diverse possano essere utilizzate in momenti diversi durante la progressione della malattia.
E non tutti gli scienziati ritengono che il ruolo del sistema immunitario nella neurodegenerazione si limiti alla microglia. Il neurologo Philip De Jager, della Columbia University di New York, sta sviluppando una terapia per l'Alzheimer basata su un bersaglio microgliale, ma afferma che anche le cellule del resto del sistema immunitario del corpo, come le cellule T, presenti in numero molto limitato nel cervello, potrebbero rivelarsi rilevanti.
Anche se l'interesse clinico sta decollando, ci sono ancora due grossi problemi: i modelli murini utilizzati nella ricerca sull'Alzheimer sono un modello grossolano per la patologia umana, ed è difficile trovare individui che siano buoni candidati per testare nuove terapie.
Alcuni di questi geni rimuovono le protezioni, o checkpoint, dalle vie che portano all'attivazione delle cellule. Altri si trovano in vie che percepiscono un danno o incoraggiano la microglia a fagocitare le molecole difettose. In ogni caso, gli schemi di espressione genica indicano che le cellule della microglia stanno incrementando le loro “pulizie di casa” per cercare di proteggere il cervello.
Le mutazioni di una dozzina circa di questi geni erano già state identificate come fattori di rischio per l'Alzheimer negli esseri umani, dice Ido Amit, immunogeneticista del Weizmann Institute of Science di Rehovot, in Israele, che ha condotto uno degli studi sugli schemi di espressione genica.
Amit dice che le cellule sono lì per una ragione e potrebbero quindi essere sfruttate. "I risultati sembravano mandarci un messaggio forte sulla biologia del sistema", afferma. Se la microglia potesse essere indotta a svolgere le sue funzioni regolari in modo più efficiente, impedendole eventuali sforzi di pulizia eccessivi, ciò potrebbe contribuire a prevenire i sintomi della malattia piuttosto che peggiorarne il decorso.
Se c'erano ancora dubbi sull'importanza della microglia nei meccanismi della demenza – se cioè svolgesse il ruolo di buono o di cattivo – questi articoli li hanno fugati. Inoltre, la microglia potrebbe anche essere indotta ad attivarsi dall'infiammazione in altre parti del corpo.
Studi epidemiologici hanno dimostrato che l'accumulo di infezioni durante la vita aumenta il rischio di deterioramento cognitivo o di demenza in età avanzata. E all'inizio di questo mese, Jonas Neher del Centro tedesco per le malattie neurodegenerative di Tubinga, e colleghi, hanno dimostrato che provocare infiammazioni nei topi iniettando molecole chiamate lipopolisaccaridi (LPS) nel loro ventre portava a cambiamenti persistenti nell'espressione genica nella microglia del cervello, anche se le molecole stesse ne rimanevano fuori. Basse dosi di LPS hanno portato a un aumento dei livelli di beta amiloide e di placche; alte dosi hanno ridotto l’accumulo.
La microglia potrebbe essere coinvolta anche in altre malattie neurodegenerative, poiché risultati simili sono stati ottenuti in modelli di sclerosi laterale amiotrofica (SLA) e morbo di Parkinson. E le ricerche di Matteoli e altri suggeriscono che potrebbero essere implicati ancora più ampiamente nei disturbi cerebrali, come il raro disturbo dello sviluppo neurologico noto come sindrome di Rett.
Al riparo dalla tempesta
Amit sta ora discutendo con i partner industriali su come potenziare le attività di pulizia della microglia. "Questo ci permetterebbe di riattivare le nostre difese naturali quando il danno è fuori controllo", dice.
Altri temono che una maggiore attivazione di microglia nelle ultime fasi della malattia possa peggiorare le cose. "Non sappiamo ancora abbastanza della biologia", dice Oleg Butovsky, neuroimmunologo della Harvard Medical School di Boston, che ha condotto l'altro studio sull'espressione genica nella microglia e sta sviluppando biomarcatori che possano essere identificati nel cervello in diverse fasi del disturbo. Egli sostiene che non è chiaro se la microglia debba essere potenziata o soppressa, o anche se tattiche diverse possano essere utilizzate in momenti diversi durante la progressione della malattia.
E non tutti gli scienziati ritengono che il ruolo del sistema immunitario nella neurodegenerazione si limiti alla microglia. Il neurologo Philip De Jager, della Columbia University di New York, sta sviluppando una terapia per l'Alzheimer basata su un bersaglio microgliale, ma afferma che anche le cellule del resto del sistema immunitario del corpo, come le cellule T, presenti in numero molto limitato nel cervello, potrebbero rivelarsi rilevanti.
Anche se l'interesse clinico sta decollando, ci sono ancora due grossi problemi: i modelli murini utilizzati nella ricerca sull'Alzheimer sono un modello grossolano per la patologia umana, ed è difficile trovare individui che siano buoni candidati per testare nuove terapie.
I topi con mutazioni genetiche che li predispongono all'Alzheimer sviluppano alcuni sintomi realistici, ma troppo rapidamente. Ciò mette i ricercatori in difficoltà quando si tratta d'identificare il momento in cui il trattamento dev'essere somministrato. "I nostri modelli sono troppo accelerati", afferma Marco Colonna della Washington University School of Medicine di St. Louis, che ha lavorato a lungo sulla biologia di TREM2. "La comunità di ricerca in questo campo riconosce che lo sviluppo di un modello in cui l'amiloide si accumula più naturalmente è una priorità."
Un'altra sfida è identificare i soggetti abbastanza precocemente nella progressione della loro malattia perché un qualsiasi farmaco sperimentale abbia la possibilità di funzionare.
I ricercatori che studiano l'Alzheimer ritengono che molti dei lavori precedenti non abbiano avuto successo non perché la loro ipotesi – che la beta amiloide e la tau siano coinvolte nella malattia in modo cruciale – sia sbagliata, ma perché il trattamento è somministrato troppo tardi.
Generalmente i pazienti vengono reclutati nelle sperimentazioni solo dopo che l'accumulo di placche e la neurodegenerazione sono progredite e la malattia probabilmente è irreversibile. Questo potrebbe anche essere uno dei motivi per cui le sperimentazioni di farmaci antinfiammatori come il naprossene o il rofecoxib hanno avuto lo stesso esito di altri trattamenti potenziali e non hanno mostrato alcun beneficio sui soggetti con il morbo di Alzheimer, dice Heneka.
I biomarcatori per identificare le persone che si trovano in una fase molto precoce della malattia si stanno rendendo disponibili solo ora. Nonostante ciò, i test sono molto costosi e complessi, e richiedono scansioni cerebrali e prelievi spinali. E hanno ancora bisogno di essere definitivamente validati nella pratica.
Le molte incertezze non stanno smorzando l'entusiasmo. "Sono stati anni eccitanti", afferma De Jager. I ricercatori del settore vedono un parallelo con l'immunoterapia del cancro, in cui il sistema immunitario viene stimolato ad attaccare i tumori. "Sembra che le malattie non ritenute classicamente immunologiche possano effettivamente avere una base immunologica".
Quando Heneka ripensa ai suoi esperimenti inaspettatamente brillanti con i topi, è cautamente ottimista sul fatto che le terapie basate sul sistema immunitario possano funzionare sul morbo di Alzheimer. Ma i nuovi studi devono affrontare i problemi che hanno afflitto le ricerche precedenti. Nessuno, dice, vuol vedere fallire l'approccio per le ragioni sbagliate. D’altra parte, non aveva mai visto un topo che si pensava avesse l'Alzheimer superare il test di memoria a pieni voti.
Le molte incertezze non stanno smorzando l'entusiasmo. "Sono stati anni eccitanti", afferma De Jager. I ricercatori del settore vedono un parallelo con l'immunoterapia del cancro, in cui il sistema immunitario viene stimolato ad attaccare i tumori. "Sembra che le malattie non ritenute classicamente immunologiche possano effettivamente avere una base immunologica".
Quando Heneka ripensa ai suoi esperimenti inaspettatamente brillanti con i topi, è cautamente ottimista sul fatto che le terapie basate sul sistema immunitario possano funzionare sul morbo di Alzheimer. Ma i nuovi studi devono affrontare i problemi che hanno afflitto le ricerche precedenti. Nessuno, dice, vuol vedere fallire l'approccio per le ragioni sbagliate. D’altra parte, non aveva mai visto un topo che si pensava avesse l'Alzheimer superare il test di memoria a pieni voti.
ENGLISH
An increasing number of studies are highlighting the crucial role of the immune system in dementia and neurodegeneration, stimulating therapeutic projects to reduce the inflammation that, together with the accumulations of beta amyloid and tau proteins, represents a characteristic signature of the disease.
Neuroscientist Michael Heneka knows that radical ideas require convincing data. In 2010, very few colleagues shared his belief that the brain's immune system played a crucial role in dementia. So in May of that year, when a series of new results provided the most stringent evidence he had ever seen of his theory, he wanted to feel excited. Instead he felt nervous.
Heneka and her group had eliminated a key inflammation gene from a strain of mice that usually develops symptoms of Alzheimer's disease. The modified mice seemed perfectly healthy. They passed tests of memory and barely showed signs of protein plaques that are a hallmark of the disease. Yet Heneka knew that her colleagues would consider the results too good to be true.
Even he was surprised at how good the rats were; he would have expected the removal of the gene, known as Nlrp3, to protect their brains, but not to approach the prevention of dementia symptoms. "I thought something had gone wrong with experimentation," says Heneka from the German Center for Neurodegenerative Diseases in Bonn. He re-analyzed the results several times. It was past midnight when he finally concluded that they could be corrected.
In the two following years, he established that nothing had gone wrong in the experiments. Together with his colleagues, he replied and elaborated the results. Since then, numerous studies have reinforced the idea of a link between dementia and the brain's immune system, highlighting the cells and signals involved. But no one has managed to identify that link unequivocally: the link seems elusive and dynamic, able to change with the progression of the disease.
However, the idea has attracted the interest of pharmaceutical investors, who have glimpsed a broad and completely unexplored market: about 50 million people worldwide suffer from dementia, a number that according to the World Health Organization will rise to 82 million by 2030. Four of the eight drug research projects supported by the Dementia Consortium - a group of charitable associations and pharmaceutical companies based in the United Kingdom that has spent 4.5 million pounds (about 5.1 million euros) ) to projects - they are focused on inflammation.
But there are obstacles. Scientists do not agree yet whether the immune system should be stimulated or inhibited depending on the different stages of the disease. And some of the practical problems that have persecuted clinical trials of Alzheimer's - imperfect mouse models and difficulties in recruiting patients at a fairly early stage of the disease - can also plague this new approach.
To weigh on this research field as dark clouds is the fact that all the clinical trials on Alzheimer's disease so far have failed. But the bioinformat Martin Hofmann-Apitius of the Fraunhofer Institute for the algorithms and the scientific calculation of Sankt Augustin, specialized in pharmaceutical research, observes that the researchers have filed numerous patents related to the objectives related to inflammation. "Soon we will see a wave of clinical trials", he predicts.
Clogged and swollen
The German psychiatrist Alois Alzheimer was the first to describe the symptoms and pathology of dementia at the beginning of the 20th century. Looking at the microscope the brain of a woman whose cognitive decline had witnessed, saw - and drew diligently - the plaques, which we now know contain the amyloid beta protein, and the fibrillary clusters of a protein called tau which, together, are the signature characteristic of the disease.
In those early representations of the diseased brain tissue, Alzheimer also designed microglia, a type of brain immune cell, nested close to neurons. "Alzheimer himself noticed the cells and drew them in abundant numbers associated with neurons," says Heneka.
Although the sketches did not establish a deeper connection between microglia and disease, Heneka remembered them when, in the mid-nineties, the ties between inflammation and Alzheimer's began to emerge. He had been fascinated by some epidemiological observations showing that people taking certain anti-inflammatory drugs (to treat rheumatoid arthritis, for example) seemed at lower risk of developing Alzheimer's than the general population.
It was encouraged by studies that microglia thickens around plaques and areas of brain degeneration and inflammatory molecules such as cytokines are concentrated in the cerebrospinal fluid of patients. Most researchers hypothesized that those observations reflected a passive response to tissue damage. But Heneka suspected that inflammation could actively provoke the disease.
Microglia has proved to be central to the link between inflammation and neurodegeneration (see the "Nature" infographic).
Its cells have two main functions. They take care of the general health of neurons and their synapses, the junctions between the neurons that allow them to communicate. And they patrol the brain, looking for threats and problems. When they detect an infectious or aberrant molecule such as beta amyloid - or debris from damaged cells - they activate and signal to other cells of the microglia to join them in an effort to clear and clean.
Some microglial proteins gather in large complexes called inflammasomes (a key component of the inflammosome is Heneka's NLRP3 protein), which produce cleaning signals in the form of activated immune molecules. Inflammasomes usually diminish once the work is finished, but in Alzheimer's they appear to remain activated, continuing to produce inflammatory molecules without being able to adequately complete the cleaning.
In 2013, microglia began to appear persistently in research on Alzheimer's disease. At about the same time that Heneka's paper showed that preventing inflammation removed Alzheimer's disease in mice, the "New England Journal of Medicine" published two large studies on the gene variants associated with the disease. Both studies linked the risk of developing late-onset Alzheimer's to a gene called TREM2, which produces a protein found in the microglial cell membrane.
Neuroscientists have begun to notice us. And so did the immunologists. Thus an interdisciplinary community of neuroimmunologists was born. "Suddenly, enormous opportunities have opened up", says neuroscientist Michela Matteoli of the University of Milan, who now runs a neuroscience program in the immunology department of the nearby Humanitas Institute, where she found a treasure of murine models lacking specific elements of the immune system, which immunologists had never had reason to use to study brain function. "Many of the tools we need are available," he says.
Good and bad
How does microglia, which has evolved to keep the brain in good condition, become a force of evil in Alzheimer's?
Last year, Heneka and colleagues published evidence suggesting a plausible mechanism for that transformation, at least in mice. They found that activated microglia cells discarded the remains of inflammasomes in small groups called granules, and that these granules continue to seed new amyloid beta clusters, spreading the disease throughout the brain. "A perfect storm," says Heneka. "Toxic amyloid beta promotes inflammation, which in turn promotes more beta amyloid toxicity."
Heneka is collaborating with immunologist Eicke Latz, from the University of Bonn, to develop a drug that inhibits the formation of inflammasome. This would allow the microglia to continue the other important roles in brain cleansing without having to resort to other microglia cells. The storm would be held at bay.
In 2016, Latz co-founded the IFM Therapeutics start-up in Boston. The company, acquired by pharmaceutical company Bristol Myers Squibb last year, already has some drug candidates that prevent the formation of inflammasomes and the hope of Latz and Heneka is to begin clinical trials in the next two years.
Meanwhile, neuroimmunologists around the world are trying to gain a deeper understanding of microglia's biology, to understand if there are other ways to design immunotherapies for Alzheimer's and other neurodegenerative diseases. Some scientists think that the healthful activities of microglia could be strengthened to eliminate the toxic beta amyloid more efficiently and avoid the storm altogether.
Two studies on post-mortem human mice and brains have shown that microglia thickens around plaques in the brain is a very specific subgroup. These cells express some genes at higher or lower levels than normal microglia, and these patterns tell an interesting story: cells seem to try to calibrate their normal cleaning tasks in order to fight plaques.
Some of these genes remove the protections, or checkpoints, from the pathways leading to cell activation. Others find themselves in ways that perceive damage or encourage microglia to engulf defective molecules. In any case, gene expression patterns indicate that microglia cells are increasing their "house cleaning" to try to protect the brain.
The mutations of about a dozen of these genes had already been identified as risk factors for Alzheimer's in humans, says Ido Amit, immunogeneticist of the Weizmann Institute of Science in Rehovot, Israel, who conducted one of the studies on gene expression.
Amit says the cells are there for a reason and could therefore be exploited. "The results seemed to send us a strong message about the biology of the system," he says. If the microglia could be induced to perform its regular functions more efficiently, preventing any excessive cleaning efforts, this could help to prevent the symptoms of the disease rather than worsen the course of the disease.
If there were still doubts about the importance of microglia in the mechanisms of dementia - if it played the role of good or bad - these articles dispelled them. Furthermore, microglia could also be induced to activate from inflammation in other parts of the body.
Epidemiological studies have shown that the accumulation of infections during life increases the risk of cognitive impairment or dementia in old age. Earlier this month, Jonas Neher of the German Center for Neurodegenerative Diseases in Tübingen, and colleagues, showed that causing inflammation in mice by injecting molecules called lipopolysaccharides (LPS) into their abdomen led to persistent changes in gene expression in the microglia of the brain, even if the molecules themselves remained outside. Low doses of LPS have led to increased levels of beta amyloid and plaques; high doses have reduced the accumulation.
Microglia may also be involved in other neurodegenerative diseases, as similar results have been obtained in models of amyotrophic lateral sclerosis (ALS) and Parkinson's disease. And the research by Matteoli and others suggests that they could be implicated even more widely in brain disorders, such as the rare neurodevelopmental disorder known as Rett syndrome.
Protected from the storm
Amit is now discussing with the industrial partners on how to improve microglia cleaning activities. "This would allow us to reactivate our natural defenses when the damage is out of control," he says.
Others fear that increased microglia activation in the last stages of the disease may make things worse. "We do not know enough about biology yet," says Oleg Butovsky, a neuroimmunologist at Harvard Medical School in Boston, who conducted the other gene expression study in microglia and is developing biomarkers that can be identified in the brain at different stages of the disorder. He argues that it is not clear whether microglia should be enhanced or suppressed, or even if different tactics can be used at different times during disease progression.
And not all scientists believe that the role of the immune system in neurodegeneration is limited to microglia. Neurologist Philip De Jager, of Columbia University in New York, is developing a therapy for Alzheimer's based on a microglial target, but also states that the cells of the rest of the body's immune system, such as T cells, are present in many limited in the brain, may prove to be relevant.
Although the clinical interest is taking off, there are still two major problems: the mouse models used in Alzheimer's research are a gross model for human disease, and it is difficult to find individuals who are good candidates for testing new therapies.
Mice with genetic mutations that predispose them to Alzheimer's develop some realistic symptoms, but too quickly. This puts researchers in difficulty when it comes to identifying the time when treatment should be given. "Our models are too fast," says Marco Colonna of the Washington University School of Medicine in St. Louis, who has worked extensively on the biology of TREM2. "The research community in this field recognizes that the development of a model in which amyloid accumulates more naturally is a priority."
Another challenge is to identify subjects early enough in the progression of their disease because any experimental drug has the ability to function.
Researchers who study Alzheimer's believe that many of the previous works have not been successful, not because their hypothesis - that beta amyloid and tau are involved in the disease in a crucial way - is wrong, but because the treatment is administered too late.
Patients are generally recruited in trials only after plaque accumulation and neurodegeneration have progressed and the disease is probably irreversible. This could also be one of the reasons why trials of anti-inflammatory drugs like naproxen or rofecoxib have had the same outcome as other potential treatments and have not shown any benefit on subjects with Alzheimer's disease, says Heneka.
Biomarkers to identify people who are at a very early stage of the disease are only becoming available now. Despite this, the tests are very expensive and complex, and require brain scans and spinal draws. And they still need to be definitively validated in practice.
The many uncertainties are not dampening enthusiasm. "They were exciting years," says De Jager. Industry researchers see a parallel with cancer immunotherapy, in which the immune system is stimulated to attack tumors. "It seems that diseases not considered classically immunological can actually have an immunological basis".
When Heneka rethinks her unexpectedly brilliant experiments with mice, she is cautiously optimistic that immune-based therapies can work on Alzheimer's. But new studies must address the problems that have plagued previous research. Nobody, he says, wants to see the approach fail for the wrong reasons. On the other hand, he had never seen a mouse that was thought to have Alzheimer's passing the memory test with flying colors.
Da:
http://www.lescienze.it/news/2018/05/19/news/fuoco_amico_cervello_malattia_di_alzheimer-3986467/?ref=nl-Le-Scienze_25-05-2018



{kind=link}
Commenti
Posta un commento