Controls in Flow of Flow Cytometry / Controlli nel flusso in citometria a flusso
Controls in Flow of Flow Cytometry / Controlli nel flusso in citometria a flusso
Segnalato dal Dott. Giuseppe Cotellessa / Reported by Dr. Giuseppe Cotellessa
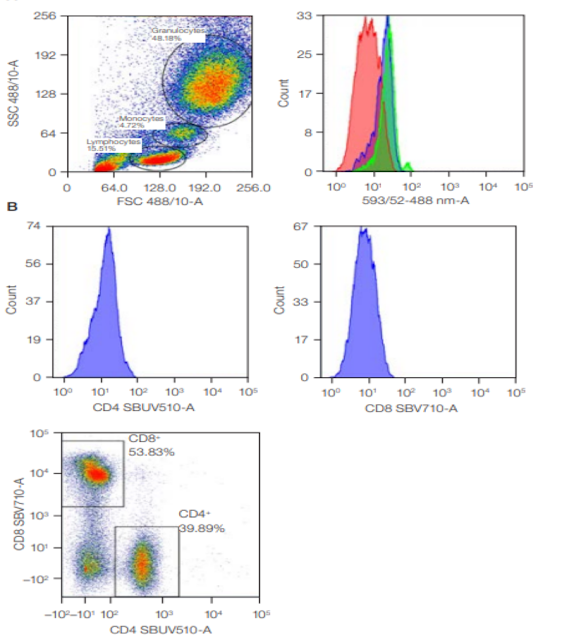
Fig. 1. Unstained controls. A, unstained peripheral blood was used to set the FSC and SSC to visualize the cells of interest and show the autofluorescence in the three marked populations (histogram key: red, lymphocytes; blue, monocytes; green, granulocytes). B, unstained cells (histograms) were used to set the PMT voltages for fluorescence channels so that fully stained cells (in the dot plot) can be evaluated. FSC, forward scatter; PMT, photomultiplier tubes; SBUV, StarBright UltraViolet; SBV, StarBright Violet; SSC, side scatter / Controlli non colorati. Un sangue periferico non colorato è stato utilizzato per impostare FSC e SSC per visualizzare le cellule di interesse e mostrare l'autofluorescenza nelle tre popolazioni contrassegnate (chiave dell'istogramma: rosso, linfociti; blu, monociti; verde, granulociti). B, le cellule non colorate (istogrammi) sono state utilizzate per impostare le tensioni PMT per i canali di fluorescenza in modo che le cellule completamente colorate (nel dot plot) possano essere valutate. FSC, dispersione in avanti; PMT, tubi fotomoltiplicatori; SBUV, StarBright UltraViolet; SBV, StarBright Violet; SSC, dispersione laterale
Controls are vital in any experiment to reliably distinguish your results from background variation and nonspecific effects. Some controls are also specific to flow cytometry and should be included in every experiment. These include unstained controls, viability controls, and compensation controls. Here, we discuss some essential controls for flow cytometry that will help you obtain publication-quality data.
Unstained Controls
One of the first things to identify in flow cytometry is your cell population. Use unstained cells to set up your instrument so that all your cells can be easily visualized on an FSC and SSC plot. Unstained cells can also be used to set your PMT voltage so that you can distinguish dim signals from autofluorescence and electronic noise while keeping the brightest cells within the scale. Historically, this was done by altering the PMT voltage so that the unstained cells lie within the first log decade for each fluorescent parameter used in your experiment (Figure 1). Using the data from unstained cells (Figure 1B), signals from stained cells can be distinguished from autofluorescence. More recently, an alternative method for setting the PMT voltages, called voltration, has gained popularity. This method uses beads with multiple fluorescence intensities to set the PMT voltage at their optimum sensitivity. The voltage is increased incrementally, and the optimal voltage is where the largest difference between the two peaks with the lowest fluorescence intensities is observed. Using this method, the negative population is not necessarily located within the first log decade and very bright signals could appear off scale. Therefore, some adjustments may be required for each experiment, but they should not be made between samples within an experiment.
Isotype Controls
In flow cytometry, background levels of staining can be a problem, especially with rare populations, cells with low expression levels, and when building multicolor panels. Isotype controls are antibodies raised against an antigen not found on the cell type or sample analyzed. Developed for surface staining, their role is to confirm the observed staining is due to a specific antibody binding to the target rather than an artifact. They should not be used to determine positive versus negative cells or to set gates, and may not be suitable for intracellular staining.
An isotype control will:
■ Determine the nonspecific binding of an antibody to Fc receptors found on monocytes, macrophages, and dendritic and B cells (Fc receptors can be blocked to reduce nonspecific binding, explained later in this chapter)
■ Confirm the observed staining is due to specific binding rather than an artifact
■ Reveal other nonspecific binding of the antibody or fluorophores, such as R-phycoerythrin (RPE or PE) and FITC (Takizawa et al. 1993, Hulspas et al. 2009), to cellular components
The most appropriate isotype control matches the:
■ Host species
■ Immunoglobulin (Ig) subclass
■ Fluorescent dye on the primary antibody If you are using a mouse IgG1 monoclonal antibody that is conjugated to FITC, you should select a mouse IgG1 isotype control conjugated to FITC. As the number of fluorophore molecules conjugated to each antibody (called the F/P ratio) differs between suppliers, it is best to purchase the isotype control from the same supplier as the primary antibody. It is also advisable, if possible, to use isotype controls at the same concentration as the primary antibody. Nonspecific antibody binding can be reduced by:
■ Blocking Fc receptors
■ Adding excess protein such as bovine serum albumin (BSA) to your buffer
■ Titrating your antibody
■ Gating out dead cells using a live/dead cell marker
■ Performing extra washing steps after staining Isoclonic Controls
One alternative to an isotype control is the isoclonic control. This is where cells are stained in the presence of an excess of identical unlabeled antibody. The latter takes up all the binding sites, preventing the labeled antibody from binding specifically. Thus, any signal that is detected must come from nonspecific binding.
Viability Controls
Dead cells have a high level of autofluorescence and nonspecific antibody binding, which can lead to false positives, a reduction in the detectable dynamic range, and a loss of resolution. This will make it difficult to detect weakly positive samples and rare populations. Using a forward and side scatter gate will allow you to exclude debris and some dead cells, but it will not remove them all. Therefore, a viability dye should be included in your flow cytometry panel. It illustrates an example in murine bone marrow, where combining a viability dye with a forward and side scatter gate significantly improves the data quality. By using a forward and side scatter gate to exclude dead cells, you can identify myeloid cells positive for both GR-1 and CD11b (upper right quadrant) in murine bone marrow. When a viability dye is introduced, in this case DAPI, you can see that the same forward and side scatter contains both live and dead cells.
When the viability dye and forward and side scatter gate are combined, the staining for CD11b and GR-1 is clearer and some staining, presumably from dead cells, disappears. There are two types of viability dyes. Nucleic acid binding dyes, such as propidium iodide, 7-AAD, and DAPI, fluoresce upon binding to nucleic acid, but cannot pass through an intact cell membrane. Therefore, only dead cells, with a permeable membrane, will fluoresce. The second type of viability dye, protein binding dyes, covalently bind to free primary amines on proteins, which are present on the surface of cells. When the membrane is compromised, the dyes permeate the cells and react with intracellular primary amines. Greater fluorescence is observed in dead cells due to their increased content of accessible free amines, allowing them to be easily distinguished from live cells.
VivaFix Cell
Viability Assays are fixable viability dyes, available in a wider range of excitation and emission spectra than nucleic acid binding dyes, for convenient analysis and addition to multicolor flow cytometry panels. Single staining will reveal the level of spectral overlap between different fluorescent dyes and allow you to remove or compensate for this overlap. As the addition of each new fluorophore can have an effect on the existing fluorophores in a panel, this spectral overlap should be compensated for every fluorophore used. Here are some important rules to remember when using single-stained samples for compensation:
1. The staining of the compensation control must be as bright as or brighter than the sample to replicate any spillover in your experiment. Antibody capture beads can be substituted for cells, and one fluorophore conjugated antibody for another, as long as the fluorescence measured is brighter for the control. The exceptions to this are tandem dyes, which cannot be substituted due to varying levels of FRET.
Note: Although it would seem safe to assume that all tandem dyes created with the same donor and acceptor would have the same emission, this is not the case. Tandem dyes from different vendors or different batches must be treated like separate dyes, and a separate single-stained control should be used for each because the amount of spillover may be different for all of these dyes.
2. The compensation algorithm needs to be performed with a positive population and a negative population. If you have a sample that is 100% positive (for example, CD45 in peripheral blood), unstained cells can be spiked into the sample to provide a negative cell population. Whether each individual compensation control contains beads, the cells used in the experiment, or even different cells, the control itself must contain particles with the same level of autofluorescence. The entire set of compensation controls may include individual samples of either beads or cells, but the individual samples must have the same carrier particles (cells or beads) as the fluorophores.
3. The compensation control must use the same fluorophore as the sample. For example, both GFP and FITC emit mostly green photons, but have vastly different emission spectra. Thus, you cannot use one of them for the sample and the other for the compensation control.
4. Enough events must be collected for the software to make a statistically significant determination of spillover. About 5,000 events for both the positive and negative populations is ideal, but fewer can be used if necessary.
5. Compensation can be applied during or after acquisition. Once you have set the PMT voltages and have applied compensation, do not change the voltages, as this will invalidate the compensation. Whenever you apply compensation, it is best to avoid doing so manually. Modern software, such as FCS Express, has automated compensation, which is much more accurate than manual methods.
Fc Blocking Controls
Fc receptors are found on monocytes, macrophages, dendritic cells, and B cells. As the name suggests, they bind antibodies via their constant Fc domain rather than the antigen specific fragment antigen-binding (Fab) domain. This nonspecific binding can lead to false positives and meaningless data. To prevent this, Fc blocking reagents (for example, Human Fc Seroblock and Murine Fc Seroblock) block Fc receptors and ensure that only antigen-specific binding is observed (Figure 2). An alternative to specialized blocking reagents is diluted serum from the same sample type (for example, mouse serum, if staining mouse cells). If used in excess, immunoglobulins in the serum will compete for the Fc receptors, preventing binding of the conjugated antibodies.

Fluorescence Minus One Controls
Fluorescence minus one (FMO) controls are important
when building multicolor flow cytometry panels, as they
will help you determine where your gates should be set.
Fluorophore spread occurs when you have multiple
fluorophores compensated in a panel. Examples of the
levels of spread of the positive population can be seen.
The spread increases with the number and brightness of the fluorophores used. Although careful experimental design, avoiding channels with a large amount of spreading, and antibody titration will help reduce this influence, FMO controls are still important. FMO controls are the experimental cells stained with all the fluorophores minus one fluorophore. It shows how the fluorescence spread from other channels can affect the data, ensuring you can position your gates accordingly. FMO controls should be included for all the fluorophores in your panel when starting a new multicolor experiment. This will then allow you to assess the spread of all the fluorophores into your missing channel and set your gates accordingly
Intracellular Staining Controls
Intracellular staining can be more problematic than surface staining, often due to higher levels of background within cells caused by protein-protein interactions. Because it requires fixation and permeabilization, which can affect antigen detection, autofluorescence, fluorophore brightness, and cell morphology, other controls are necessary. Controls such as a negative cell line, an antibody known to be negative on your cells, or a secondary antibody alone (if using primary and secondary antibodies) can be useful to determine specific binding in intracellular staining. An example of using these controls in intracellular staining can be seen in Figure 3.

Fig. 3. Intracellular controls. Cas9-positive (red) or Cas9-negative (green) HEK-293T cells were stained with an Anti-Cas9 Antibody and a secondary antibody conjugated to FITC (#STAR121F). An unstained control (gray) and a secondary-only control (blue) were included. FITC, fluorescein isothiocyanate. / Controlli intracellulari. Le cellule HEK-293T Cas9-positive (rosse) o Cas9-negative (verdi) sono state colorate con un anticorpo Anti-Cas9 e un anticorpo secondario coniugato a FITC (#STAR121F). Sono stati inclusi un controllo non colorato (grigio) e un controllo solo secondario (blu). FITC, isotiocianato di fluoresceina.
Using an internal control within your sample is a good example of control for intracellular staining. When staining for a T cell–specific marker in peripheral blood, check that the B cells and monocytes are negative within the same sample. Additionally, because intracellular staining requires fixation and permeabilization, staining without permeabilizing the cells will give you some information on the levels of background surface staining. As well as controlling for the antibodies, appropriate controls for the other reagents must also be included. The fixative and permeabilization buffers used for intracellular staining may affect your cells’ morphology, so you may have to alter the position of the gates normally used in analysis. The fixation and permeabilization conditions may need optimizing and some reagents, such as the fluorophore APC-Cy7, may not be suitable.
Biological Controls
In addition to staining and isotype controls, you should also consider biological controls that will enable you to determine staining specificity and experimental limitations. They are important for all staining but especially intracellular staining, which can have higher background fluorescence. Controls include known negative samples and known positive samples. These include cells known to either express or lack expression of the antigen of interest, where the antigen has been knocked down or out using RNA interference (RNAi) or CRISPR technology to produce a negative cell, or that have been transfected and the antigen is being overexpressed to verify positive staining. For some experiments, such as cytokine release measurement, both an unstimulated and a fully stimulated sample is important to determine both positive results and the dynamic range of fluorescence staining, and to confirm the antibody is performing as expected. An example of this can be seen, in which human peripheral blood lymphocytes were stained for CD154 and CD3 on stimulated and unstimulated cells. The pattern of antibody staining matches what would be predicted from these cell types, indicating that the antibodies are working as intended. Using these antibodies, positive samples can be distinguished from negative, demonstrating that the antibody has an appropriate dynamic range. This type of control may also help you choose the most appropriate fluorophore, as small changes may have better resolution with brighter fluorophores. Understanding your experiment and sample is important when choosing the right biological control.
ITALIANO
I controlli sono fondamentali in qualsiasi esperimento per distinguere in modo affidabile i risultati dalla variazione di sfondo e dagli effetti non specifici. Alcuni controlli sono anche specifici per la citometria a flusso e dovrebbero essere inclusi in ogni esperimento. Questi includono controlli non colorati, controlli di fattibilità e controlli di compensazione. Qui, discutiamo alcuni controlli essenziali per la citometria a flusso che ti aiuteranno a ottenere dati di qualità della pubblicazione.
Controlli non colorati
Una delle prime cose da identificare nella citometria a flusso è la popolazione cellulare. Usa le cellule non colorate per configurare il tuo strumento in modo che tutte le tue cellule possano essere facilmente visualizzate su un grafico FSC e SSC. Le cellule non colorate possono anche essere utilizzate per impostare la tensione PMT in modo da poter distinguere i segnali deboli dall'autofluorescenza e dal rumore elettronico mantenendo le cellule più luminose all'interno della scala. Storicamente, ciò è stato fatto alterando la tensione PMT in modo che le cellule non colorate si trovino entro la prima decade logaritmica per ciascun parametro fluorescente utilizzato nell'esperimento (Figura 1). Utilizzando i dati delle cellule non colorate (Figura 1B), i segnali delle cellule colorate possono essere distinti dall'autofluorescenza. Più recentemente, ha guadagnato popolarità un metodo alternativo per impostare le tensioni PMT, chiamato voltaggio. Questo metodo utilizza perline con più intensità di fluorescenza per impostare la tensione PMT alla loro sensibilità ottimale. La tensione viene aumentata in modo incrementale e la tensione ottimale è dove si osserva la maggiore differenza tra i due picchi con le intensità di fluorescenza più basse. Utilizzando questo metodo, la popolazione negativa non si trova necessariamente entro la prima decade logaritmica e segnali molto luminosi potrebbero apparire fuori scala. Pertanto, potrebbero essere necessarie alcune regolazioni per ciascun esperimento, ma non dovrebbero essere apportate tra campioni all'interno di un esperimento.
Controlli isotipici
Nella citometria a flusso, i livelli di fondo della colorazione possono essere un problema, specialmente con popolazioni rare, cellule con bassi livelli di espressione e quando si costruiscono pannelli multicolori. I controlli isotipici sono anticorpi prodotti contro un antigene non trovato sul tipo cellulare o sul campione analizzato. Sviluppati per la colorazione della superficie, il loro ruolo è quello di confermare che la colorazione osservata è dovuta ad un anticorpo specifico che si lega al bersaglio piuttosto che ad un artefatto. Non devono essere utilizzati per determinare cellule positive rispetto a quelle negative o per impostare i gate e potrebbero non essere adatti per la colorazione intracellulare.
Un controllo isotipico:
■ Determinare il legame non specifico di un anticorpo ai recettori Fc presenti su monociti, macrofagi e cellule dendritiche e B (i recettori Fc possono essere bloccati per ridurre il legame non specifico, spiegato più avanti in questo capitolo)
■ Confermare che la colorazione osservata è dovuta a un legame specifico piuttosto che a un artefatto
■ Rivelare altri legami non specifici dell'anticorpo o dei fluorofori, come R-ficoeritrina (RPE o PE) e FITC (Takizawa et al. 1993, Hulspas et al. 2009), ai componenti cellulari
Il controllo isotipico più appropriato corrisponde a:
■ Specie ospite
■ Sottoclasse di immunoglobuline (Ig).
■ Colorante fluorescente sull'anticorpo primario Se si utilizza un anticorpo monoclonale IgG1 murino coniugato a FITC, è necessario selezionare un controllo isotipico IgG1 murino coniugato a FITC. Poiché il numero di molecole di fluoroforo coniugate a ciascun anticorpo (chiamato rapporto F/P) varia tra i fornitori, è meglio acquistare il controllo isotipico dallo stesso fornitore dell'anticorpo primario. È inoltre consigliabile, se possibile, utilizzare controlli isotipici alla stessa concentrazione dell'anticorpo primario. Il legame anticorpale non specifico può essere ridotto da:
■ Blocco dei recettori Fc
■ Aggiunta di proteine in eccesso come l'albumina sierica bovina (BSA) al tampone
■ Titolazione dell'anticorpo
■ Eliminazione delle cellule morte utilizzando un marcatore di cellule vive/morte
■ Esecuzione di ulteriori fasi di lavaggio dopo la colorazione dei controlli isoclonici
Un'alternativa ad un controllo isotipico è il controllo isoclone. È qui che le cellule vengono colorate in presenza di un eccesso di anticorpo identico non marcato. Quest'ultimo occupa tutti i siti di legame, impedendo all'anticorpo marcato di legarsi in modo specifico. Pertanto, qualsiasi segnale rilevato deve provenire da un legame non specifico.
Controlli di vitalità
Le cellule morte hanno un alto livello di autofluorescenza e legame anticorpale non specifico, che può portare a falsi positivi, una riduzione dell'intervallo dinamico rilevabile ed una perdita di risoluzione. Ciò renderà difficile rilevare campioni debolmente positivi e popolazioni rare. L'uso di un cancello di dispersione anteriore e laterale ti consentirà di escludere detriti ed alcune cellule morte, ma non li rimuoverà tutti. Pertanto, un colorante di vitalità dovrebbe essere incluso nel pannello di citometria a flusso. Esso illustra un esempio nel midollo osseo murino, in cui la combinazione di un colorante di vitalità con un gate di dispersione anteriore e laterale migliora significativamente la qualità dei dati. Utilizzando una porta di dispersione in avanti e laterale per escludere le cellule morte, è possibile identificare le cellule mieloidi positive sia per GR-1 che per CD11b (quadrante in alto a destra) nel midollo osseo murino. Quando viene introdotto un colorante di vitalità, in questo caso DAPI, puoi vedere che la stessa dispersione in avanti e laterale contiene sia cellule vive che morte.
Quando il colorante di vitalità ed il gate di dispersione anteriore e laterale vengono combinati, la colorazione per CD11b e GR-1 è più chiara e alcune macchie, presumibilmente dalle cellule morte, scompaiono. Esistono due tipi di coloranti di vitalità. I coloranti che legano gli acidi nucleici, come ioduro di propidio, 7-AAD e DAPI, emettono fluorescenza quando si legano all'acido nucleico, ma non possono passare attraverso una membrana cellulare intatta. Pertanto, solo le cellule morte, con una membrana permeabile, emetteranno fluorescenza. Il secondo tipo di colorante di vitalità, i coloranti che legano le proteine, si legano in modo covalente alle ammine primarie libere sulle proteine, che sono presenti sulla superficie delle cellule. Quando la membrana è compromessa, i coloranti permeano le cellule e reagiscono con le ammine primarie intracellulari. Una maggiore fluorescenza si osserva nelle cellule morte a causa del loro maggiore contenuto di ammine libere accessibili, che consentono loro di essere facilmente distinte dalle cellule vive. I test di vitalità cellulare VivaFix sono coloranti di vitalità fissabili, disponibili in una gamma più ampia di spettri di eccitazione ed emissione rispetto ai coloranti leganti l'acido nucleico, per una comoda analisi e aggiunta a pannelli di citometria a flusso multicolori. La colorazione singola rivelerà il livello di sovrapposizione spettrale tra i diversi coloranti fluorescenti e consentirà di rimuovere o compensare questa sovrapposizione. Poiché l'aggiunta di ogni nuovo fluoroforo può avere un effetto sui fluorofori esistenti in un pannello, questa sovrapposizione spettrale deve essere compensata per ogni fluoroforo utilizzato. Ecco alcune regole importanti da ricordare quando si utilizzano campioni con colorazione singola per la compensazione:
1. La colorazione del controllo di compensazione deve essere brillante o più brillante del campione per replicare qualsiasi spillover nell'esperimento. Le sfere di cattura dell'anticorpo possono essere sostituite con le cellule ed un anticorpo coniugato con fluoroforo con un altro, purché la fluorescenza misurata sia più luminosa per il controllo. Fanno eccezione i coloranti tandem, che non possono essere sostituiti a causa dei diversi livelli di FRET. Nota: sebbene sembrerebbe lecito ritenere che tutti i coloranti tandem creati con lo stesso donatore e accettore avrebbero la stessa emissione, non è così. I coloranti in tandem di diversi fornitori o lotti diversi devono essere trattati come coloranti separati e per ciascuno deve essere utilizzato un controllo a colorazione singola separata perché la quantità di spillover può essere diversa per tutti questi coloranti.
2. L'algoritmo di compensazione deve essere eseguito con una popolazione positiva e una popolazione negativa. Se si dispone di un campione positivo al 100% (ad esempio CD45 nel sangue periferico), le cellule non colorate possono essere aggiunte al campione per fornire una popolazione cellulare negativa. Indipendentemente dal fatto che ogni singolo controllo di compensazione contenga sfere, cellule utilizzate nell'esperimento o anche cellule diverse, il controllo stesso deve contenere particelle con lo stesso livello di autofluorescenza. L'intera serie di controlli di compensazione può includere singoli campioni di perline o cellule, ma i singoli campioni devono avere le stesse particelle di supporto (cellule o perline) dei fluorofori.
3. Il controllo di compensazione deve utilizzare lo stesso fluoroforo del campione. Ad esempio, sia GFP che FITC emettono principalmente fotoni verdi, ma hanno spettri di emissione molto diversi. Pertanto, non è possibile utilizzarne uno per il campione e l'altro per il controllo della compensazione.
4. È necessario raccogliere un numero sufficiente di eventi affinché il software esegua una determinazione statisticamente significativa dello spillover. Circa 5.000 eventi sia per le popolazioni positive che per quelle negative sono l'ideale, ma se necessario è possibile utilizzarne meno.
5. Il risarcimento può essere applicato durante o dopo l'acquisizione. Dopo aver impostato le tensioni PMT e aver applicato la compensazione, non modificare le tensioni, poiché ciò invaliderà la compensazione. Ogni volta che si applica un compenso, è meglio evitare di farlo manualmente. I software moderni, come FCS Express, hanno una compensazione automatizzata, che è molto più accurata dei metodi manuali.
Controlli di blocco Fc
I recettori Fc si trovano su monociti, macrofagi, cellule dendritiche e cellule B. Come suggerisce il nome, legano gli anticorpi tramite il loro dominio Fc costante piuttosto che il dominio di legame dell'antigene del frammento specifico dell'antigene (Fab). Questo legame non specifico può portare a falsi positivi e dati privi di significato. Per evitare ciò, i reagenti di blocco Fc (ad esempio, sieroblocco Fc umano e sieroblocco Fc murino) bloccano i recettori Fc e assicurano che venga osservato solo il legame antigene-specifico (Figura 2). Un'alternativa ai reagenti bloccanti specializzati è il siero diluito dello stesso tipo di campione (ad esempio siero di topo, se si colorano le cellule di topo). Se usate in eccesso, le immunoglobuline nel siero competono per i recettori Fc, impedendo il legame degli anticorpi coniugati.
5. La compensazione può essere applicata durante o dopo l'acquisizione. Dopo aver impostato le tensioni PMT ed aver applicato la compensazione, non modificare le tensioni, poiché ciò invaliderà la compensazione. Ogni volta che si applica un compenso, è meglio evitare di farlo manualmente. I software moderni, come FCS Express, hanno una compensazione automatizzata, che è molto più accurata dei metodi manuali.
Controlli di blocco Fc
I recettori Fc si trovano su monociti, macrofagi, cellule dendritiche e cellule B. Come suggerisce il nome, legano gli anticorpi tramite il loro dominio Fc costante piuttosto che il dominio di legame dell'antigene del frammento specifico dell'antigene (Fab). Questo legame non specifico può portare a falsi positivi e dati privi di significato. Per evitare ciò, i reagenti di blocco Fc (ad esempio, sieroblocco Fc umano e sieroblocco Fc murino) bloccano i recettori Fc e assicurano che venga osservato solo il legame antigene-specifico (Figura 2). Un'alternativa ai reagenti bloccanti specializzati è il siero diluito dello stesso tipo di campione (ad esempio siero di topo, se si colorano le cellule di topo). Se usate in eccesso, le immunoglobuline nel siero competono per i recettori Fc, impedendo il legame degli anticorpi coniugati.
Fluorescenza meno uno controlli
I controlli della fluorescenza meno uno (FMO) sono importanti quando si costruiscono pannelli di citometria a flusso multicolori, poiché ti aiuteranno a determinare dove dovrebbero essere impostati i tuoi gate. La diffusione dei fluorofori si verifica quando si hanno più fluorofori compensati in un pannello. Esempi dei livelli di diffusione della popolazione positiva possono essere visti.
La diffusione aumenta con il numero e la luminosità dei fluorofori utilizzati. Sebbene un'attenta progettazione sperimentale, evitando i canali con una grande quantità di diffusione e la titolazione degli anticorpi contribuiranno a ridurre questa influenza, i controlli FMO sono ancora importanti. I controlli FMO sono le cellule sperimentali colorate con tutti i fluorofori meno un fluoroforo. Esso mostra come la diffusione della fluorescenza da altri canali può influenzare i dati, assicurando di poter posizionare i gate di conseguenza. I controlli FMO dovrebbero essere inclusi per tutti i fluorofori nel pannello quando si avvia un nuovo esperimento multicolore. Ciò ti consentirà quindi di valutare la diffusione di tutti i fluorofori nel canale mancante e di impostare i gate di conseguenza
Controlli della colorazione intracellulare
La colorazione intracellulare può essere più problematica della colorazione superficiale, spesso a causa di livelli più elevati di sfondo all'interno delle cellule causati dalle interazioni proteina-proteina. Poiché richiede fissazione e permeabilizzazione, che possono influenzare il rilevamento dell'antigene, l'autofluorescenza, la luminosità del fluoroforo e la morfologia cellulare, sono necessari altri controlli. Controlli come una linea cellulare negativa, un anticorpo noto per essere negativo sulle cellule o un anticorpo secondario da solo (se si utilizzano anticorpi primari e secondari) possono essere utili per determinare il legame specifico nella colorazione intracellulare. Un esempio di utilizzo di questi controlli nella colorazione intracellulare può essere visto nella Figura 3.
L'uso di un controllo interno all'interno del campione è un buon esempio di controllo per la colorazione intracellulare. Quando si esegue la colorazione per un marcatore specifico delle cellule T nel sangue periferico, verificare che i linfociti B e i monociti siano negativi all'interno dello stesso campione. Inoltre, poiché la colorazione intracellulare richiede fissazione e permeabilizzazione, la colorazione senza permeabilizzare le cellule fornirà alcune informazioni sui livelli di colorazione della superficie di fondo. Oltre al controllo degli anticorpi, devono essere inclusi anche i controlli appropriati per gli altri reagenti. I tamponi fissativi e di permeabilizzazione utilizzati per la colorazione intracellulare possono influenzare la morfologia delle cellule, quindi potrebbe essere necessario modificare la posizione dei gate normalmente utilizzati nell'analisi. Potrebbe essere necessario ottimizzare le condizioni di fissazione e permeabilizzazione e alcuni reagenti, come il fluoroforo APC-Cy7, potrebbero non essere adatti.
Controlli biologici
Oltre alla colorazione ed ai controlli isotipici, dovresti anche considerare i controlli biologici che ti consentiranno di determinare la specificità della colorazione ed i limiti sperimentali. Sono importanti per tutte le colorazioni, ma soprattutto per le colorazioni intracellulari, che possono avere una fluorescenza di fondo maggiore. I controlli includono campioni negativi noti e campioni positivi noti. Questi includono cellule note per esprimere o meno l'espressione dell'antigene di interesse, in cui l'antigene è stato abbattuto o eliminato utilizzando l'interferenza dell'RNA (RNAi) o la tecnologia CRISPR per produrre una cellula negativa, o che sono state trasfettate e l'antigene viene sovraespresso per verificare la colorazione positiva. Per alcuni esperimenti, come la misurazione del rilascio di citochine, sia un campione non stimolato che uno completamente stimolato sono importanti per determinare sia i risultati positivi che l'intervallo dinamico della colorazione di fluorescenza e per confermare che l'anticorpo si comporta come previsto. Un esempio di ciò può essere visto, in cui i linfociti del sangue periferico umano sono stati colorati per CD154 e CD3 su cellule stimolate e non stimolate. Il modello di colorazione degli anticorpi corrisponde a ciò che sarebbe previsto da questi tipi di cellule, indicando che gli anticorpi funzionano come previsto. Utilizzando questi anticorpi, i campioni positivi possono essere distinti da quelli negativi, dimostrando che l'anticorpo ha un intervallo dinamico appropriato. Questo tipo di controllo può anche aiutarti a scegliere il fluoroforo più appropriato, poiché piccole modifiche possono avere una risoluzione migliore con fluorofori più luminosi. Comprendere l'esperimento e il campione è importante quando si sceglie il giusto controllo biologico.
Da:
https://offers.the-scientist.com/hubfs/TS_PPL_Bio-Rad%20EU_Evergreen%20Flow%20Cytometry_Guide_406213/flow-cytometry-basics-guide.pdf?hsCtaTracking=ae192164-1471-4fb9-9577-ce6d1cadc0e5%7Ccdaea2a5-8e3e-43c3-8d2c-a82844d2313f



Commenti
Posta un commento